 電子發燒友App
電子發燒友App


目前已商業化的鋰離子電池電極材料中的過渡金屬存在溶解等交叉效應,嚴重影響著電池的循環性能。然而,當前關于交叉效應的研究大都基于氧化物正極的半電池,對氧化物正極和鋰金屬負極電池中交叉化學物質的影響知之甚少。
在本工作中,作者在具有高鎳正極、鋰金屬負極和局部高濃度電解液(LHCE)的電池中探索了正極到負極和負極到正極交叉的影響。研究表明與高鎳正極配對的鋰金屬負極的固體電解液界面(SEI)生長量是與鋰金屬配對的鋰金屬負極的三倍。同時,與鋰金屬配對的正極的容量衰減比與石墨配對的相同正極高2-3倍。其中FSI鹽的分解和交叉被確定為這些變化的主要來源。
【內容詳情】
為了區分一個電極對另一個電極的影響,作者用不同的電極組合組裝了三組軟包電池,包括具有高鎳LiNi0.94Co0.06O2正極(NC9406或僅NC)和鋰金屬負極(Li)的電池(記作:NC|Li)、具有高鎳正極和石墨負極(Gr)的電池(記作:NC|Gr)以及鋰對稱電池(記作:Li|Li)。
1. 電池和電極性能
如圖1所示,具有LHCE、NC9406正極和鋰金屬負極的軟包電池在200次循環后仍保持84%的容量。具有石墨負極的電池的容量保持率甚至更高,達到93%。與NC|Gr電池相比,NC|Li電池的容量衰減增加可歸因于負極或正極與負極的相互作用(即交叉效應)。
通常考慮到鋰金屬負極的高反應性,人們直觀地認為鋰金屬負極對NC|Li電池的容量衰減有很大貢獻。然而,如圖1c的對稱電池數據所示,與直覺相反,鋰金屬負極通常在循環過程中表現出最小的阻抗增長。相反,一旦鋰金屬或電解液幾乎耗盡,鋰金屬負極的反應性通常表現為快速失效。為了弄清本研究中鋰金屬負極在對稱電池中的阻抗增長最小,但與NC9406配對時阻抗增長顯著的現象,作者拆開循環的軟包電池,并用循環電極的穿孔組以及新鮮的電解液和對電極裝紐扣電池。通過這種方式,可以獨立測量每個循環電極的容量和過電位。
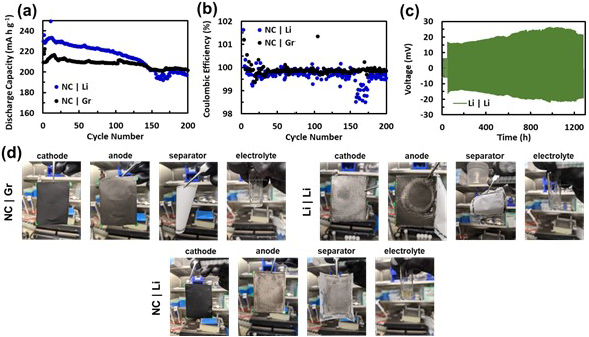
圖1. 具有局部高濃度電解液的軟包電池的循環性能和拆卸。(a) NC|Li和NC|Gr電池在循環過程中的比容量和(b)庫侖效率;(c) Li|Li過循環的電壓曲線;(d) 200次循環后循環電極、隔膜和電解液的圖像。
有趣的是,與Li|Li的電池相比,具有NC|Li的紐扣電池最初確實具有降低的容量(圖2b,2c)。這可能表明針對NC循環的鋰金屬負極確實比在對稱電池中循環的阻抗增長更多。然而,這些紐扣電池的容量在5-10個周期內達到平衡,并且它們的電壓曲線重疊良好,表明阻抗相似。盡管循環鋰金屬負極的早期潤濕可能存在一些差異,這些重新組裝的紐扣電池可比較的過電位和穩態容量表明NC|Li的鋰金屬負極對該電池的容量衰減沒有貢獻。因此,NC|Li電池相對于NC|Gr更大的衰減歸因于正極。與循環正極組裝的紐扣電池表明,具有NC|Li正極的紐扣電池在第一次循環中的容量明顯低于具有NC|Gr正極的紐扣電池。與具有循環鋰的紐扣電池不同,這種較低的容量也與增加的過電勢相吻合,如圖2c所示,較低的容量在整個循環過程中保持不變。此外,~18 mA h g-1的容量差異與NC|Li和NC|Gr軟包電池的衰減差異相當合理,而NC|Li軟包電池的最大容量比NC|Gr高約17 mAh g-1,最終容量比NC|Gr低約5 mA h g-1,變化量約為22 mA h g-1.
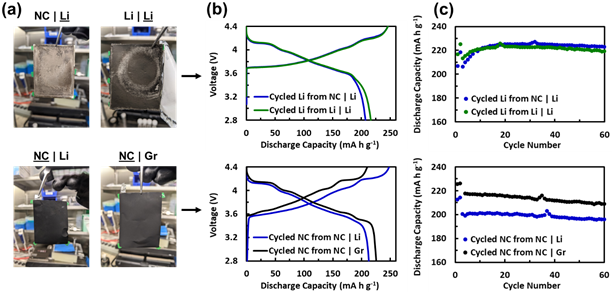
圖2. 用新鮮電解液、新鮮對電極組裝的紐扣電池和從拆卸的軟包電池中回收的循環電極的循環性能。(a)顯示用于重新組裝電池的電極的圖像;(b)電壓曲線和(c)重新組裝電池的循環數據。
2. 正極的體積和表面表征
如前所述,兩個相同正極的容量衰減差異一定是由于不同負極的交叉效應造成的。要了解交叉對容量衰減的作用,有必要確定衰減的物理起源。就塊狀晶體結構而言,兩個正極似乎都相對不受循環的影響。這在意料之中,因為高鎳正極通常在幾百個循環的過程中有最小的陽離子混合。雖然已知高鎳正極會隨著循環時間的延長而破裂,但這種現象通常在超過200次循環時也能觀察到。此外,最近的研究表明,高穩定性電解液(如LHCE)通過抑制應力腐蝕開裂機制進一步減輕了開裂和微開裂,并且預計開裂導致該系統的容量衰減。
如果循環后的NC|Li的形態和晶體學變化很少,那么它的容量衰減與表面現象有關。為了探索這一點,作者用X射線光電子能譜(XPS,圖3)分析了循環后的正極。正如預期的那樣,每個正極都顯示出來自溶劑分解的碳質物質以及來自FSI陰離子分解的含氮、硫和氟物質。有趣的是,對于與鋰金屬配對的正極,特別是氮和硫的正極,一些鹽分解峰更為明顯。雖然這可能表明NC|Li電池中的鹽分解更大,但與鹽分解和LiF形成相關的氟峰的強度反而降低了。同時,NC|Li的S 2p光譜不僅顯示出硫的增加,而且還存在一些與元素硫和適度氧化的S-O相關的獨特峰。相對于FSI的雙氧合硫,這些硫物質被還原是出乎意料的。雖然與鹽分解相關的氟、硫和氮光譜在NC|Li和NC|Gr之間存在顯著差異,但碳和氧光譜相對沒有變化。最顯著的差異是在533 eV附近的O 1s的峰值,這歸因于包括S-O、N-O和C-O在內的各種物種。雖然這些很難分成單獨的峰,但具有更多硫和氮氧化物的樣品應該具有更高強度的混合峰是合理的。
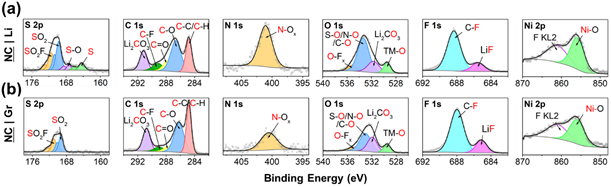
圖3. 循環200次后循環NC9406正極的XPS數據:(a)正極與鋰配對,(b)正極與石墨配對。
為了進一步探索循環正極的表面特性,作者還用飛行時間二次離子質譜(TOF-SIMS)對正極進行了分析。圖4給出了NC|Li和NC|Gr正極的一些代表性分子片段的深度剖面,其中每個片段都歸一化為其自身的最大強度。該數據中最引人注目的觀察結果是以62Ni-為代表的鎳信號的強度。該信號來自大塊正極材料,該信號達到最大值的80%所需的時間/深度是衡量正極-電解液界面(CEI)厚度的指標。NC|Li電池的CEI約為40 nm,而NC|Gr電池的CEI約為140 nm。該結果與傳統觀點相反,經歷了2-3倍容量衰減的正極的CEI薄了約3倍。為了證實這一結果的真實性,作者在同一樣品的不同區域重復測量兩次,并獲得了相似的結果。在多次測量中看到的NC|Gr稍微“變薄”的CEI可歸因于較短的濺射時間,這種情況下不允許62Ni-信號達到穩態。
綜上所述,雖然NC|Li電池的CEI看起來比NC|Gr電池薄幾倍,但是成分的變化似乎抵消了更薄CEI的好處。特別是與鋰配對的正極(NC|Li)比與石墨配對的正極(NC|Gr)具有更大比例的硫和氮,而氟的比例更小。對于硫,這種成分差異部分表現為存在僅在NC|Li上發現的還原硫物質(S, S-O)。就有機CEI成分而言,樣品之間的差異似乎很小。因此,鹽分解物種似乎至少最初是交叉效應的主要驅動力。
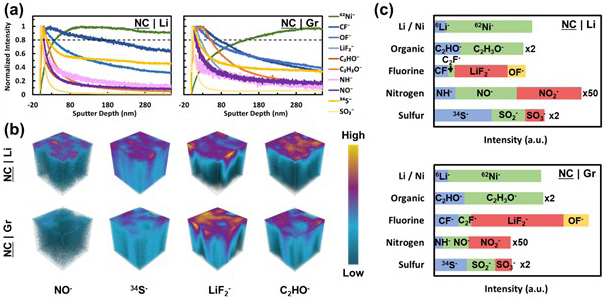
圖4. 循環NC9406正極在200次循環后的TOF-SIMS數據。(a) 深度剖面顯示幾個特征分子片段的歸一化強度;(b) 顯示幾個片段分布的3D圖,樣品面積100 x 100 μm,高度為350 nm;(c) 各種碎片的總綜合強度,選擇的碎片按比例放大以提高可見度。
3. 鋰金屬負極的體積和表面表征
按理說,如果鋰金屬負極影響正極,那么鋰金屬負極本身也應該受到影響。前面的結果已經表明,NC|Li和Li|Li電池的循環負極的阻抗增長在200次循環內是相當的。因此,有必要對鋰金屬負極SEI的物理特性進行表征。圖5顯示了循環鋰金屬負極的軸上和橫截面SEM圖像,揭示了SEI厚度的顯著差異。盡管它們相似且有限的阻抗增長,NC|Li上的SEI比Li|Li上的SEI薄三倍。應該注意的是,本文中的“SEI”是指SEI組件和死鋰的復雜框架,這是循環鋰金屬負極的典型特征。除了厚度之外,NC|Li的SEI形態也更致密且分辨率更好。這些圖像是在放電(鋰剝離)條件下拍攝的,因此應該更能表明SEI成分而不是鋰金屬形態。考慮到這一點,Li|Li樣品的混濁及其缺乏明顯的微觀結構可能是由于較高比例的無定形有機物質所致。為了證實這一點并澄清SEI組成,作者再次使用XPS對Li|Li樣品進行了表征。
為了避免由于NC|Li的鋰金屬負極出現充電問題對XPS測量準確性的影響,作者選擇循環5次后的金屬負極進行XPS測試,結果如圖6所示。由于5個循環可能不足以讓交叉物種達到穩定狀態,因此該數據應被視為確定交叉效應的“最壞情況”。其中,NC|Li電池的氮和硫信號相對于Li|Li大幅減少了約40%。但是無論如何,氮和硫與氟的比例仍然在質量上符合預期:相對于氟,NC|Li電池上氮和硫的富集與NC|Li上的相反行為相關。

圖5. (a) 循環鋰金屬負極在200次循環后的橫截面和(b)軸上SEM圖像,每個循環有兩個放大倍數。
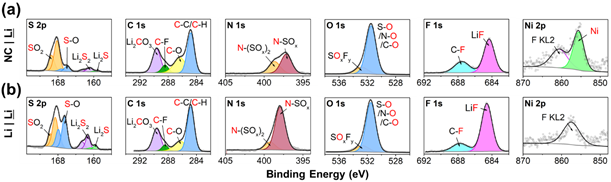
圖6. 5次循環后循環鋰金屬負極的XPS數據:(a) 鋰金屬負極與正極配對,(b) 鋰與鋰配對。
在循環鋰金屬負極上硫和氮的鍵合環境中也可以看到一些明顯的趨勢。雖然Li|Li電池的硫含量通常高于NC|Li電池,但這種額外的硫主要處于與更多還原物質相關的較低結合能,例如Li2S、Li2S2和適度氧化的S-O。與循環正極相比,這很有趣,之前已經討論過NC|Li電池具有更大比例的還原硫物質,例如S-O和元素硫。這可能與來自負極的還原物質的清除有關,這也可以解釋相對于Li|Li,NC|Li電池上這些物質的含量較低。這可以描述為多硫化物穿梭的一種形式。在這種機制中,FSI中的一部分硫首先在鋰金屬負極上脫去氧,最終導致低階硫化物,如Li2S和Li2S2。在電池放電期間,這些硫化物轉化為可溶于DME溶劑的高級多硫化物,就像在鋰-硫電池中一樣。在NC|Li電池中,這些多硫化物穿過正極,在那里它們被氧化成元素硫和硫氧化物。與鋰-硫電池不同,正極電壓永遠不會低到足以將硫還原成多硫化鋰,因此“多硫化物穿梭”是單向且不可逆的。石墨,沒有金屬鋰容易還原SO2,形成這些硫化鋰的速度較慢,因此循環NC|Gr電池沒有測到元素硫。
NC|Li和Li|Li電池上氮的鍵合環境也不同(圖6)。NC|Li電池上的平均結合能和氧化態低于Li|Li。在N 1s中看到的這種更大比例的N-(SOx)鍵與在S 2p中看到的SO2相對于S-O的更高濃度相匹配。顯然,鹽在NC|Li上的分解不太徹底,或者那些分解程度更高的分子通過交叉去除。在有機物種方面,兩個鋰金屬負極樣品非常相似,無法分離各種氧物種。雖然NC|Li的碳譜似乎確實相對于Li|Li有適度的CO-O增加,但其碳譜在其他方面是相同的。C-O鍵的增加可歸因于多種機制,例如由富氟SEI驅動的溶劑分解差異,或來自正極的氧化溶劑種類的交叉。
因此,關于鋰金屬負極的主要結論反映了循環正極的結論。NC|Li的SEI/死鋰層厚度是Li|Li上厚度的三分之一,其中SEI厚度是穩定性的直接衡量標準,因為它代表了活性鋰的損失。NC|Li相對于氟富含硫和氮,而NC|Li相對于硫和氮富含氟(分別與NC|Gr和Li|Li相比)。這可以解釋為FSI中的氟在硫和氮穿梭到正極之前被鋰金屬負極優先清除,反之亦然。此外,作者還發現了單向多硫化物穿梭的證據,這進一步耗盡了負極中的硫并使其在正極上富集。作者在循環鋰金屬負極(和循環石墨)上發現了過渡金屬沉積物,盡管它們似乎不太可能促成SEI厚度減少3倍。與正極一樣,溶劑交叉效應對鋰金屬負極的影響很小。
4. 電解液分析和交叉機制
為了進一步研究哪些物種可能參與了交叉,作者從循環電池中回收電解液并使用傅里葉變換紅外光譜(FTIR)和核磁共振光譜(NMR)對其進行表征,結果表明電解液的整體組成幾乎保持不變。
最終,FSI鹽陰離子被提議作為該系統中交叉效應的主要驅動因素,甚至超出了其作為多硫化物穿梭的硫源的作用。與其他機制相比,基于FSI的交叉解釋了更多觀察到的表面征,并且鑒于已知鹽分解在濃縮電解液中占主導地位,因此是直觀的。該機制可以描述如下:在鋰金屬負極上,FSI分解的第一步是除氟。在鋰對稱電池中,產生的脫氟FSI在兩個電極上形成,并繼續在電解液中積聚。最終,脫氟的FSI開始與鋰金屬反應,將其氮和硫添加到鋰SEI中。同時,在具有鋰金屬負極和高鎳正極的電池中,脫氟FSI無法在電解液中積聚。相反,它會越過正極,在那里脫氟FSI較低的氧化穩定性導致其快速消耗。這種脫氟FSI與正極的交叉減少了鋰金屬SEI中的硫和氮,并允許FSI連續脫氟,從而增加氟含量。正極受到相反的影響,硫和氮增加,氟含量減少。正極表面成分的這些變化通過帶有石墨負極的電池得到證實,與鋰金屬負極相比,石墨負極向正極輸送的脫氟FSI物質更少。
鑒于氟作為SEI保護成分的已知狀態,交叉對氟含量的影響可以解釋鋰金屬負極與高鎳正極配對時的出色穩定性。同樣,氟含量的降低可以解釋正極在同一系統中穩定性較差的原因。硫和氮的含量也可能起作用,盡管這不能與氟的影響分開。雖然基于FSI的交叉在所研究的系統中似乎是有害的,但必須考慮到本研究中使用了過量的鋰和電解液,在鋰稀薄的系統中,跨接實際上可以通過保護鋰金屬負極來延長循環壽命,而鋰金屬負極通常是鋰金屬電池中的限制電極。這項工作還進一步證明,無論是通過添加劑還是更重氟化的鹽,SEI和CEI中的氟含量越高,可以實現循環壽命的提高。但是,添加劑或鹽改性對溶解度、熱穩定性等仍有嚴格的要求。最后,必須注意的是,在具有多重交叉效應的系統中,很難完全分得清因果關系。因此,該系統還有很大的進一步研究和優化空間。
審核編輯:湯梓紅














 工商網監
工商網監
評論